
Portal Hypertension
Share
Follow Us
Increased portal vein pressure caused by intrinsic liver disease or obstruction in the extrahepatic portal vein or hepatic venous outflow tract.
The portal venous system carries all blood from the abdominal GI tract, spleen, pancreas, and gallbladder back to the heart through the liver. The portal vein is formed by the union of the superior mesenteric and splenic veins. At the porta hepatis it divides into the right and left branches, which are segmentally distributed intrahepatically; the terminal portal venules drain into the sinusoids. In the resting state, the portal vein carries about 1 to 1.2 L/min of blood (about 75% of total hepatic blood flow) and provides 2/3 of the liver’s O2 supply. The portal vein is valveless; thus, pressure in the portal system depends on the product of input from blood flow in the portal vein and total hepatic resistance to outflow. Normal pressure in the portal vein is between 1 and 4 mm Hg higher than the free pressure in the hepatic vein, or up to 6 mm Hg higher than right atrial pressure. Portal hypertension is defined as pressures above these limits.
Pathogenesis
Portal pressure can be defined by the equation P (portal pressure) = Q (blood flow in the portal venous system) x R (hepatic resistance). Any condition causing increased portal venous flow, or increased hepatic resistance, can develop into portal hypertension. In practice, most conditions associated with portal hypertension are due to a combination of these 2 factors.
In the West, cirrhosis of any etiology is the most common cause of portal hypertension. In the cirrhotic liver, architectural disorganization with nodular regeneration and fibrosis results in a large increase in resistance due to vascular destruction and distortion. The normally smooth, regular vascular channels become tortuous and irregular, which also increases resistance. Further splanchnic arterial inflow and thus flow in the portal vein also increase. Although it appears that increased resistance is the prime factor in cirrhosis, increased flow also contributes to portal hypertension.
Rare conditions causing purely an increased flow into the portal venous system are almost always associated with only mild portal pressure elevation. However, secondary structural changes in the liver microcirculation lead to increased resistance (eg, a splanchnic arteriovenous [A-V] shunt may develop after abdominal trauma). Although significant portal hypertension with this condition has been documented, when the liver is carefully examined in detail, subtle but significant microcirculatory changes are present (eg, collagen deposition in the space of Disse). Conditions associated purely with increased portal venous flow (eg, massive splenomegaly and splanchnic A-V malformations and shunts) are associated with significant portal hypertension only in the presence of secondary intrahepatic microcirculatory changes.
Symptoms, Signs, and Diagnosis
Most clinical consequences of portal hypertension can be attributed to the development of portosystemic collateral vessels to return splanchnic blood to the heart. These vessels may form at several sites in the gut circulation. The most important are esophageal varices, formed by gross dilation of esophageal submucosal veins. These vessels carry blood from the coronary veins of the portal system into the azygos-hemiazygos veins. Other collateral sites include the umbilical vein into the omphalomesenteric vein. This occasionally results in striking dilation and prominence of the collateral vessels on the anterior abdominal wall with centrifugal radiation outward from the umbilicus, a pattern known as caput medusae. Rarely, a venous hum may be heard over such dilated veins (Cruveilhier’s sign). Other sites of collateral formation include the retroperitoneal cavity, splenorenal veins between the left kidney and the spleen, and vessels between the rectal and inferior mesenteric circulations. The latter results in large dilated, inferior rectal veins, often mistaken as gross hemorrhoids. Rarely, collaterals can develop in atypical sites (eg, duodenum, colon, or vagina). Portopulmonary collaterals have also been described.
These collateral vessels result in shunting of portal blood into the systemic circulation, causing high systemic concentrations of several hormones and substances normally extracted by the liver. The pathogenesis of hepatic or portosystemic encephalopathy is often ascribed to a failure of the liver to degrade a putative metabolic toxin produced in the gut.
Since cirrhosis is the dominant cause of portal hypertension in the West, presentation will be that of cirrhosis with decompensation, or directly attributable to portal hypertension. This includes GI bleeding, ascites and edema, encephalopathy, or nonspecific constitutional symptoms (eg, fatigue, lethargy, anorexia).
Radiologic investigations may provide clues to the presence of portal hypertension. A plain x-ray of the abdomen may show a ground-glass appearance due to ascites and an enlarged spleen. US of the abdomen may show ascites, abnormal density and texture of the liver, and occasionally dilated portal veins and collaterals, if they are large. Doppler US can determine blood flow, patency, and caliber of the portal vein. Esophageal varices may be recognized as a tortuous, worm-like appearance of the mucosa on a barium swallow, but are best directly visualized during endoscopy. Radionuclide liver scan often shows a patchy uptake in the liver associated with increased bone marrow and splenic uptake. CT scans of the abdomen can identify dilation of the portal vein and often collateral vessels (eg, the azygos vein). Venography, either indirectly (venous phase of a celiac axis angiogram especially with subtraction techniques) or directly (splenic venography or transhepatic portography), visualizes the portal system to identify venous occlusion and collateral flow.
Portal pressure can be measured by several methods. A thin needle may be introduced percutaneously directly into the hepatic parenchyma. Similarly, a thin needle may be introduced percutaneously into the spleen to measure splenic pulp pressure, an accurate reflection of splenic vein pressure. The latter technique also offers the advantage of enabling injection of contrast material into the splenic venous system, providing a splenoportogram. Another approach involves fluoroscopic guidance of a thin (Chiba) needle percutaneously and then transhepatically into a portal vein branch. Alternatively, a small-bore cannula can be introduced percutaneously or at laparotomy into the umbilical vein and, thus, into the omphalomesenteric vein, since this system becomes prominent in portal hypertension. A drawback common to all of these foregoing procedures is the lack of an internal zero reference, the zero value being estimated level of the right atrium. In addition, piercing hepatic or splenic parenchyma entails a small but significant risk of bleeding (1 to 3%). Probably the best technique involves catheterization of the hepatic vein through either a jugular or femoral vein approach. Wedging of the catheter in a small hepatic vein branch results in a pressure that closely approximates the portal pressure in most causes of liver disease. When the catheter tip lies free in the hepatic vein, this free hepatic vein pressure normally should be 1 to 4 mm Hg less than the wedged hepatic pressure. The difference between the wedge and free hepatic venous pressures, also known as the hepatic venous pressure gradient, portohepatic gradient, or corrected sinusoidal pressure, represents the contribution of the hepatic sinusoids to portal pressure.
Classification:
Portal hypertension has been subclassified traditionally according to the presumed site of resistance. Presinusoidal hypertension can be either intra- or prehepatic: Prehepatic (extrahepatic) causes include portal and splenic vein thromboses. Intrahepatic presinusoidal hypertension occurs in schistosomiasis, myelofibrosis and leukemic liver infiltration, idiopathic portal fibrosis, nodular regenerative hyperplasia, and granulomatous diseases (eg, sarcoidosis and early stages of primary biliary cirrhosis). In all cases of presinusoidal hypertension, the directly measured portal venous pressure will greatly exceed the hepatic venous pressure gradient, which should be normal or near normal. The presinusoidal block prevents transmission of the elevated portal pressure to the wedged hepatic vein. The overwhelming basis for sinusoidal and postsinusoidal portal hypertension is cirrhosis, particularly that due to alcohol. Postsinusoidal hypertension can again be divided into intrahepatic and posthepatic. Posthepatic causes include chronic heart failure, constrictive pericarditis, and obstruction of the hepatic venous outflow tract by membranous webs in the inferior vena cava. Intrahepatic postsinusoidal causes of portal hypertension include occlusive disease of the small veins and venules (veno-occlusive disease) and occlusions in large hepatic veins (Budd-Chiari syndrome). All cases of sinusoidal and postsinusoidal portal hypertension are associated with hepatic venous pressure gradients, which are about equal to the directly measured portal venous pressures. The resistance to flow extends from the hepatic venous system to the portal vein.
This traditional classification is somewhat arbitrary, since the actual site of resistance in many conditions is unclear. In practical terms, almost all presinusoidal conditions are associated with relatively well-preserved liver function, whereas the sinusoidal and postsinusoidal conditions generally have cirrhosis or otherwise deranged function. Therefore, bleeding or surgery is generally better tolerated by the patient with presinusoidal hypertension.
Prognosis and Treatment
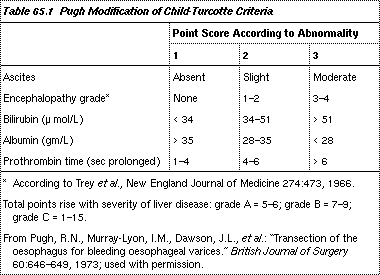
Prognosis is critically dependent on liver function. Liver failure is often the prime cause or a significant associated factor in mortality. The in-hospital (all inclusive) mortality rates with GI bleeding in Child-Pugh grade A (5 to 10%), B (15 to 25%), and C (50 to 70%) patients allow stratification of patients to improve assessment of outcome. Criteria for each grade are given in table 65.1.
Treatment:
All patients with portal hypertension who have GI hemorrhage must be hospitalized. About 50 to 75% of bleeding episodes in cirrhotic patients are a direct consequence of portal hypertension: Bleeding is from either ruptured gastroesophageal varices or diffuse oozing from the gastric mucosa (congestive gastropathy). In congestive gastropathy, mesenteric venous hypertension leads to congestion of the gastric mucosa, particularly the fundus, rendering it more susceptible to damage and bleeding even with minor insults (eg, modest alcohol or aspirin ingestion). Other causes of GI bleeding (eg, duodenal or gastric ulcer, Mallory-Weiss lacerations) account for bleeding in the remaining patients.
Standard resuscitation measures include fluid and blood transfusions. Sedatives should be avoided, in view of possible encephalopathy. In the latter, cleansing enemas will remove blood products from the bowel, and lactulose will reduce hepatic encephalopathy. To determine the site of bleeding, endoscopy within 24 h of admission is advisable.
The most difficult, lethal bleeding is caused by variceal rupture. Emergency measures to stop variceal bleeding include mechanical balloon tamponade, vasoconstrictor drugs, and particularly endoscopic sclerotherapy, which is considered the first choice of therapy. Surgery should be avoided, if possible, because of the high operative mortality rate.
Types of balloon devices for esophageal tamponade are the Minnesota tube with an esophageal suction port, the Sengstaken-Blakemore tube with both an esophageal and gastric balloon, and the Linton-Nachlas tube with only a large gastric balloon. In practice, all are equally effective but potentially dangerous. Only experienced physicians should use these devices; the complication rate is 10 to 20%, even in experienced hands. Complications include aspiration pneumonia, esophageal rupture, and asphyxia.
Vasoactive agents that lower portal pressure are vasopressin (20 u. IV over 10 to 20 min) and a long-acting analog (terlipressin, and somatostatin or one of its analogs). Prolonged vasopressin infusions at lower dosage (eg, 0.1 to 0.4 u./min given over > 4 h) should be avoided due to side effects of coronary and mesenteric vasoconstriction that can lead to infarction, renal shutdown with oliguria, and local tissue necrosis if the IV infusion extravasates. Besides, long-term use is limited by the development of tachyphylaxis. Concomitant sublingual nitroglycerin (0.3 mg q 1 h) may help avoid some of the side effects of vasopressin. European experience with terlipressin suggests that its efficacy and side effects are comparable to vasopressin. Somatostatin may prove to have fewer side effects and equivalent efficacy.
The procedure of choice is endoscopic sclerotherapy. Variceal injections of several types of sclerosing agents effectively control acute bleeding and, later as elective therapy, eradicate varices once patients have stabilized. Complications of sclerotherapy include esophageal ulceration and perforation and, rarely, pulmonary embarrassment or portal vein thrombosis.
Surgery has a limited role. If bleeding is unresponsive or recurrent, the simplest, safest intervention is esophageal transection with a stapling gun. This controls bleeding in 90 to 95% of cases. A beneficial effect on survival has yet to be shown with any form of surgical therapy.
Chronic therapy:
Once the acute bleeding episode is over and the cirrhotic patient becomes stable , further treatment may be with drugs, sclerotherapy, or surgery. Propranolol appears to reduce rebleeding risk in a minority of patients, but responders cannot be easily identified, so routine use cannot yet be recommended. Other drugs (eg, clonidine, prazosin, ketanserin, and verapamil) are even more experimental.
Sclerotherapy is currently the procedure of choice, but changes in long-term prognosis remain unproven. For patients who continue to rebleed despite sclerotherapy, surgical options include liver transplantation, if indicated, or a shunt procedure to decompress the portal venous system into the systemic circulation. Various types of portacaval and mesocaval shunts, as well as the splenorenal shunts (including the distal splenorenal shunt, the Warren shunt), all divert some or all of the portal blood away from the liver. These procedures tend to precipitate hepatic encephalopathy, a disabling condition that has limited the value of portal decompression. Other side effects include deterioration of liver function and onset or progression of hepatic iron deposition (hemosiderosis).


