
Molecular Basis for Persistent Hepatitis B Virus Infection In the Liver After Clearance of Serum Hepatitis B Surface Antigen
Share
Follow Us
HEPATOLOGY, June 1998, p. 1736-1742, Vol. 27, No. 6
Andrew L. Mason1, Lizhe Xu1, Linsheng Guo1, Mary Kuhns2, and Robert P. Perrillo1
From the 1 Section of Gastroenterology and Hepatology, Ochsner Medical Institutions, New Orleans, LA; and 2 Abbott Laboratories, Abbott Park, IL.
ABSTRACT
Hepatitis B virus (HBV) DNA has been detected by polymerase chain reaction (PCR) in the liver of patients with resolved chronic HBV infection and sustained clearance of Hepatitis B surface antigen (HBsAg) from serum. However, it is unknown whether the virus is transcriptionally active at this time or if the covalently closed circular (CCC) replicative intermediate of HBV DNA can still be detected. Therefore, hepatic nucleic acid extracts from seven patients who had cleared serum HBsAg were assessed by (PCR) for either reverse-transcribed HBV RNA, or an intact direct repeat region of the HBV genome indicative of the CCC replicative intermediate of HBV DNA. HBV transcripts were detected in four of seven patients in the study group, whereas an intact direct repeat region of the HBV genome was detected in three. Evidence for viral transcription and replication was more frequently detected in patients who had recently cleared serum HBsAg, but HBV RNA was also detected in one patient 5 years after HBsAg clearance, and an intact direct repeat region of HBV DNA was detected in another subject at nearly 4 years after resolution of disease. Therefore, hepatic HBV transcription may be associated with replicative intermediates of persistent HBV DNA in patients who have cleared HBsAg from serum, suggesting that, on occasion, HBV may not be in a latent state but undergoing low-level replication. (HEPATOLOGY 1998;27:1736-1742.)
INTRODUCTION
The clearance of the Hepatitis B surface antigen (HBsAg) from serum usually indicates a resolution of biochemical and histological hepatitis in patients with chronic Hepatitis B.1 However, there are clearly documented cases of reactivation of latent viral infection following chemotherapy and immunosuppressive treatment, as well as de novo infection in patients receiving organs from HBsAg-negative donors with serological evidence of previous Hepatitis B virus (HBV) infection.2-7 In this setting, the reactivated infection is not entirely unexpected, because most patients who have cleared HBsAg from the serum still have detectable HBV DNA in the liver using the polymerase chain reaction (PCR) methodology.8-14 In fact, HBV DNA can also be found in bodily secretions and peripheral blood mononuclear cells from patients with acute and chronic HBV infection after sustained loss of serum HBsAg.15-17 Taken together, these studies suggest that following the loss of HBsAg from serum, HBV persists in a state of low-level replication or in a replication-competent state that can be reactivated to form infectious particles.
The natural biology of the latent virus may be further investigated by assessing the transcriptional activity and the molecular form of the persistent HBV DNA. Using in situ hybridization, we were unable to detect hepatic HBV RNA in patients who had cleared serum HBsAg, and other investigators had little success using reverse-transcription (RT) PCR studies because of the inability to completely eradicate tissue HBV DNA.11 Furthermore, the molecular form of the persistent HBV DNA has proved difficult to define because of the minute quantities of virus found in patients with resolved hepatitis.
During chronic infection, HBV DNA can be detected in four predominant molecular forms. Following infection by the intact Dane particle, the partially double-stranded HBV-DNA genome becomes a covalently closed circular (CCC) DNA molecule that acts as a template for transcription of mRNA and the RNA pregenome. The reverse transcription of the RNA pregenome and second-strand HBV DNA synthesis results in low-molecular-weight replicative intermediates, whereas the HBV DNA that integrates into the host’s genome can be detected as high-molecular-weight species on Southern blot.18,19 Similar to retroviral integration at the site of the long terminal repeats, HBV DNA generally inserts into DNA flanked by the direct repeat regions, which share sequence homology with the U5 region of murine leukemia virus long terminal repeats.20-22
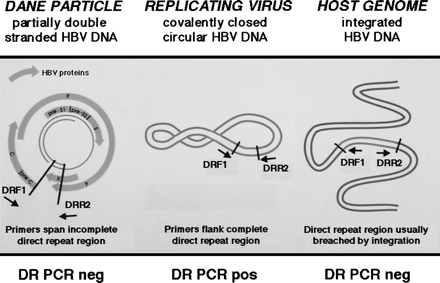
The genomic organization of the HBV direct repeat region provides a potential strategy for the detection of replicating virus when only minute quantities of HBV DNA are present (Fig. 1). Using PCR primers flanking the direct repeat region, the CCC HBV DNA can be amplified using PCR. In contrast, the HBV DNA within the Dane particle is incomplete, and integrated HBV DNA is usually, but not always, disrupted in the direct repeat region (Fig. 1). Therefore, the latter molecular forms as depicted in Fig. 1 cannot be amplified using these direct repeat PCR primers. This methodology has been previously employed to study the emergence of CCC duck HBV DNA during acute infection in vitro.23 In our study, to investigate the molecular basis for the latent HBV infection, we performed RT-PCR to assess transcriptional activity and PCR of the HBV direct repeat region to detect the CCC HBV-DNA replicative intermediate of HBV using nucleic acid extracted from the livers of patients with a remote history of chronic HBV infection.
|
Fig. 1. PCR methodology used to detect an intact direct repeat region of HBV DNA of CCC HBV DNA intermediate of replicating virus. Using DRF1 and DRR2 oligonucleotide primers spanning the direct repeat region of the HBV genome, the PCR of native viral DNA in the Dane particle will not amplify a product, because the partially double-stranded HBV DNA is disrupted in the direct repeat region. Also, this PCR methodology will not amplify a product from integrated HBV DNA that is interrupted in the direct repeat region (in an analogous fashion to retroviruses that integrate into the genome using the long terminal repeats). However, a PCR product can be amplified using the DRF1 and DRR2 primers from the replicating CCC HBV DNA, because the direct repeat region is contiguous in this molecular form. |
PATIENTS AND METHODS
Study Population. The study group consisted of 7 patients who had a documented history of chronic HBV infection for over 1 year, who then subsequently cleared serum HBsAg and serum HBV DNA (AUSZYME-II/AUSRIA-II, and HBV-DNA assay, Abbott Laboratories, Abbott Park, IL). Each patient had participated in antiviral clinical trials for chronic Hepatitis B; 2 had been treated with interferon alfa alone, 3 with prednisone withdrawal followed by either adenine arabinoside phosphate (n = 1) or interferon alfa (n = 2), and 2 patients were untreated.1 None of the patients had evidence of hepatitis delta nor Hepatitis C virus infection, but 1 patient was anti-human immunodeficiency virus-positive. Patients were followed for 3 to 67 months after disappearance of serum HBsAg, at which time a liver biopsy was obtained (table 1). Each patient was assessed by PCR for persistent HBV DNA in serum liver and peripheral blood mononuclear cells. One of 7 were HBV-DNA-positive in the serum, 6 of 7 were HBV-DNA-positive in the liver,8 and 4 of 7 were HBV-DNA-positive in the peripheral blood mononuclear cells16 at the time of the repeat biopsy.
| table 1. Southern Blot Studies of Hepatic Nucleic Acids Amplified by Either RT-PCR Using Primers Complementary to the HBV Core and Surface Genes or Seminested PCR Using Primers Flanking the HBV DR Region |
Control liver biopsies were derived from 15 HBsAg-positive patients. Three of these patients were Hepatitis B e antigen-positive, another 2 presented with fulminant hepatic failure, and the remainder were deemed to be healthy carriers. Liver biopsies from 4 patients who underwent liver transplantation for biliary atresia, alcohol-related liver disease, primary biliary cirrhosis, and primary sclerosing cholangitis, respectively, were used as negative controls. These 4 patients had no history or serological evidence of HBV infection, and HBV DNA was not detected by PCR in the total cellular DNA extracted from their native liver.24 Additional control samples included DNA extracted from the HBsAg-producing hepatoma cell line, PLC/PRF/5,22 and sucrose gradient-purified Dane particles (approximately 560 particles per PCR reaction) derived from pooled serum of patients with chronic HBV.
Extraction of Nucleic Acids From Liver. For RNA extraction, frozen liver tissues from either needle liver biopsies (5 mg to 20 mg) or liver explants (100 mg to 300 mg) were homogenized in 0.5 mL or 2.0 mL, respectively, of 4 mol/L guanidine isothiocyanate (Gibco BRL, Gaithersburg, MD), 25 mmol/L sodium citrate, 0.5% sarcosyl, and 0.72% ![]() -mercaptoethanol (Sigma, St. Louis, MO).
-mercaptoethanol (Sigma, St. Louis, MO).
After adding one-tenth volume of 3 mol/L sodium acetate, the samples were incubated for 16 hours at 37°C, vortexed for 4 minutes with an equal volume of water-saturated phenol, and for a further 4 minutes with a one-tenth volume of 24:1 chloroform/isoamyl alcohol (all reagents from Sigma). After placing the tubes on ice for 15 minutes, the samples were centrifuged at 10,000 rpm for 10 minutes, and the upper aqueous phase was then vortexed with an equal volume of chloroform/isoamyl alcohol for 4 minutes, placed on ice for 15 minutes, and centrifuged again for 10 minutes at 10,000 rpm. The upper aqueous phase was then precipitated in double the volume of RNase-free alcohol at ![]() 20°C for 2 hours, centrifuged for 30 minutes at 10,000 rpm, and resuspended in 50 to 200 µL diethyl-pyrocarbonate (Sigma)-treated water.
20°C for 2 hours, centrifuged for 30 minutes at 10,000 rpm, and resuspended in 50 to 200 µL diethyl-pyrocarbonate (Sigma)-treated water.
Total hepatic DNA was extracted from liver biopsies as previously described, resuspended at a concentration of 20 ng to 50 ng/µL in sterile water, and 200 to 500 ng of DNA was used for each PCR reaction.16
RT-PCR. The RNA detection was performed both by a one-step method using Retrotherm RTTM (Epicentre Technology, Madison, WI), which has reverse transcriptase and thermostable DNA polymerase activity, and by a two-step method using Moloney murine leukemia virus reverse transcriptase (Gibco BRL, Gaithersburg, MD), followed by PCR amplification with Taq polymerase (Perkin Elmer Cetus, Norwalk, CT).
For the one-step method, 1 to 2 µg of total hepatic RNA was added to 2.5 µL of 20× Retrotherm Reaction buffer A and 2.5 µL 20x Retrotherm Reaction buffer B (1× concentration; 10 mmol/L Tris-HCL [pH 8.3], 50 mmol/L KCl, 1.5 mmol/L MgCl2, and 0.75 MnCl2) to a final volume of 45 µL in diethyl pyrocarbonate-treated water, denatured at 95°C, cooled on ice, and then incubated at 37°C for 15 minutes with 100 units of DNase 1 (Gibco BRL, Grand Island, NY) and 20 Units RNasin (Promega, Madison, WI). Following heat denaturation of the DNase for 5 minutes at 99°C, the RNA samples were incubated with 5 units of Retrotherm, 20 units of RNasin, 100 ng of the 5′ sense and 3′ antisense oligonucleotide primers, 1 mmol/L final concentration of nucleotide mix (Promega) in a total volume of 50 µL, at 50°C for 5 minutes and 10 minutes at 70°C to synthesize the first cDNA strand, and then at 94°C for 1 minute, 50°C for 5 minutes, and 70°C for 5 minutes to synthesize the second cDNA strand.
For the two-step method, 1 to 2 µg of total hepatic RNA was added to 10 µL of Gene Amp 10× PCR buffer (Perkin Elmer Cetus) (1× concentration; 10 mmol/L Tris-HCL [pH 8.3], 50 mmol/L KCl, 1.5 mmol/L MgCl2, and 0.001% [wt/vol] gelatin) and diethyl pyrocarbonate-treated water to a final volume of 50 µL, denatured at 95°C, cooled on ice, and then incubated at 37°C for 15 minutes with 100 units of DNase 1 and 20 units RNasin. Following heat denaturation at 99°C for 5 minutes, the samples were incubated with 100 units of Moloney murine leukemia virus RT, 20 units of RNasin, 100 ng of the 3′ antisense oligonucleotide, 1 mmol/L final concentration of nucleotide mix, for 1 hour at 37°C. The samples were heat-denatured again at 99°C for 5 minutes, and the following reagents for PCR were added to the same tube: 3 units Taq polymerase, 1 mmol/L final concentration of nucleotide mix, 200 ng of the 5′ sense and 100 ng of the 3′ antisense oligonucleotide primers, and sterile water to a final volume of 100 µL.
Each sample was analyzed using PCR methodologies described previously using oligonucleotide primers complementary to the HBs gene (primers MD 13 and MD 14), Hepatitis B core antigen gene, and the albumin gene as a positive control to assess the integrity of the extracted RNA.25-27 As a control for HBV DNA contamination in the RNA samples, the 5′ sense primer was used for the reverse-transcription step in the two-step method instead of the 3′ antisense primer, and, additionally, the reverse-transcription step was omitted before the PCR amplification. Also, as a specificity control for the one-step method, each sample was pretreated with RNAse A (Sigma) to ensure that a positive signal was abrogated by RNAse pretreatment and only HBV RNA was being amplified.
Our methodology used to detect CCC HBV DNA was similar to that employed to identify duck CCC HBV DNA.23 Three oligonucleotide primers, DRF1: GTCTGTGCCTTCTCATCTGC (nucleotides 1553-1572), DRR2: ACAAGAGATGATTAGGCAGAGG (nucleotides 1830-1851), and DRR3: AGTATGGTGAGGTGAGCAATGC (nucleotides 2040-2061), were designed for this study using GeneJockeyTM software (Cambridge, UK) to flank the direct repeat region between the Hepatitis B core and polymerase genes as depicted inFig. 1. The PCR was performed using 100 ng of each primer, DRF1 and DRR3, in 50 µL reaction mix for 30 cycles at 94°C for 1 minute, 53°C for 1.5 minutes, and 72°C for 3 minutes. Seminested PCR was then performed using 2 µL of the latter PCR product with 100 ng of primers DRF1 and DRR2 in 50 µL reaction mix for 30 cycles at 94°C for 1 minute, 49°C for 1.5 minutes and 72°C for 3 minutes.
To prevent PCR product contamination, the extraction of RNA, the RT-PCR, and analysis of PCR products were performed in separate rooms by different personnel, all nonenzymatic reagents were ultraviolet-irradiated before use for 6 minutes in a UV Stratalinker 1800 (Stratagene, La Jolla, CA), and Tipguard Aerosol free pipette tips (Midwest Scientific, Valley Park, MO) were used. Four water blanks were routinely assessed with each PCR run.
Ten percent of each PCR product was run on an 0.8% agarose gel, denatured, blotted onto a Zetabind nylon filter (Cuno, Meriden, CT), cross-linked in a UV Stratalinker 1800, and then hybridized to a 3.2-kb HBV-DNA probe (cloned HBV DNA kindly provided by John Casey, Georgetown University, MD) labeled with32P by a Prime-It Random Primer Kit (Stratagene) or by a 5′ 32P-labeled internal oligonucleotide specific for each primer pair.25-27 Following high-stringency washes at 65°C for the full-length HBV-DNA probe, or at 42°C for the oligonucleotide probes, the filters were exposed to X Omat film (Kodak, Rochester, NY) for 24 hours to 1 week at ![]() 70°C.
70°C.
RESULTS
All 7 study patients and 17 of 18 control patients had detectable albumin mRNA by RT-PCR, and these samples were used for HBV-RNA detection. As these positive control primers span a 549-bp intron in the albumin gene, the inadvertent amplification of DNA results in an 822-bp molecular-weight band on an ethidium bromide-stained agarose gel, rather than the expected 273-bp molecular-weight PCR product derived from albumin mRNA.27 The Retrotherm one-step method consistently demonstrated greater intensity of the bands on the agarose gel and Southern blot than the RT followed by PCR two-step method, and the results derived from this method were used for analysis (compare results with patient 7 in Fig. 2A, using the two-step method and Fig. 2B with the one-step method).
|
Fig. 2. (A) Southern blot hybridization analyses of a 524-bp RT-PCR product amplified from total hepatic RNA from a HBsAg-positive patient (HBs) and a HBsAg-negative patient (patient 7) by the two-step method. Before PCR with oligonucleotides complementary to the Hepatitis B core antigen gene, the initial RT was performed with either the antisense primer RT/as to HBV RNA, the sense primer RT/s, or no reverse transcriptase enzyme noRT, as specificity controls. Only the RT performed with the antisense primer in lanes RT/as had demonstrable bands after autoradiographic exposure for 7 days
Fig 2B (B) Southern blot hybridization analyses of 524-bp Retrotherm amplified HBV-RNA product using oligonucleotide primers complementary to Hepatitis B core antigen in the one-step method. The total hepatic RNA was derived from an HBsAg-positive patient (HBs), and the study group patients who cleared serum HBsAg (1 to 7). In the HBsAg-negative group, 7 demonstrated the strongest reactivity when compared with the other reactive patients (1 and 20). W, water blank control. |
.gif) Fig 2A
Fig 2A2.gif)
The specificity controls demonstrated that only the antisense primers to HBV RNA used in the RT step followed by PCR amplification detected HBV nucleic acid sequences. No hybridization signal to HBV nucleic acid sequences was observed on Southern blot when either the RT step was omitted, the sense primer to HBV RNA was used for the RT step, or by the one-step method following RNAse digestion of the sample before PCR amplification (Fig. 2A). Therefore, only HBV RNA was being amplified as any residual HBV DNA in the samples could have been detected by PCR amplification after: 1) omitting the RT in the two-step method; 2) using the incorrect primer in the RT step in the two-step method; or 3) following the RNAse digestion in the one-step method. No HBV RT-PCR products were detected in the total hepatic RNA derived from the four negative control patients (table 1) nor in the water blanks incorporated into each PCR experiment (Fig. 2B).
HBV RNA was detected in the liver in 4 of 7 study group patients following clearance of HBsAg (table 1). Only HBsAg-positive patients had visible RT-PCR products on the ethidium bromide-stained agarose gel, and the HBV RNA was demonstrated by Southern blot hybridization of RT-PCR products in the HBsAg-negative study group (Fig. 2B). Patient 3 was HBV-RNA-positive in the liver using HBsAg primers but negative using the Hepatitis B core antigen primers (table 1). HBV RNA was detected 3 months after HBsAg loss in 2 patients, and 9 months after clearance of HBsAg in a third patient. In contrast to these patients, patient 7, who had cleared serum HBsAg 67 months previously, still had appreciable HBV RNA in the liver. Of note, this patient consistently had the most intense hybridization signal on Southern blot (Fig. 2B) and also was the only patient in the study group to have HBV DNA detected by PCR in the serum and liver at the time of biopsy.8 All of the HBsAg-positive patients had detectable HBV RNA in the liver, and 9 of 11 had positive hybridization signal on Southern blot when the HBsAg primers were used for RT-PCR, and 10 of 11 were positive using Hepatitis B core antigen primers (table 1).
Using the Dane particle DNA as a template, PCR products were detected using both the HBc gene and HBs gene primers, but no reactivity was observed with the DR primers, demonstrating specificity for double-stranded DNA in the direct repeat region of the HBV genome (Fig. 1). Using the latter seminested PCR of the DR region, PCR products were observed in 3 of 7 study group patients, the DNA extracted from the PLC/PRF/5 cell line, and the HBsAg-positive control patients (Fig. 3). The samples of 2 of these patients, who also had detectable HBV RNA, were collected 3 and 9 months following loss of serum HBsAg, whereas the third patient was biopsied 46 months’ post-HBsAg clearance and at that interval had no evidence of viral transcription using RT-PCR (table 1). Of note, patients 2 and 7, with detectable viral transcription 3 months and 67 months following loss of serum HBsAg, respectively, had no evidence of CCC HBV DNA using the DR region seminested PCR methodology (table 1).
|
Fig. 3. Southern blot hybridization of analysis of a 298-bp PCR product using primers spanning the DR region of HBV genome demonstrating positive signal in HBs-positive control and three study group patients (1, 3, and 5). W, water blank control. |
.gif)
DISCUSSION
This study was performed in follow-up to our last report documenting HBV DNA in the liver and serum of our patients following loss of serum HBsAg.8Previously, we suggested that low-level HBV replication may be occurring in the liver, but the source of serum HBV DNA, from liver or extrahepatic reservoirs, and the molecular form of HBV DNA within the liver, either integrated, episomal, or virion DNA, remained unexplored.8 The findings of this study now demonstrate that patients who recover from chronic Hepatitis B infection and clear HBsAg from serum may still harbor an intact direct repeat region of HBV DNA, found in the CCC HBV DNA (Fig. 1), as well as transcriptionally active virus in the liver. While the data suggesting ongoing low-level viral replication were mainly derived from patients who had recently lost serum HBsAg, evidence for viral transcription or a complete direct repeat region was also observed at remote intervals following HBsAg clearance. For example, more than 5 years after clearance of HBsAg, viral transcription was detected in the liver of patient 7, who, in a previous PCR study, still had serum HBV DNA in the serum,8 and an intact direct repeat region of HBV DNA was found in patient 5 at nearly 4 years after HBsAg loss. In addition, we observed both HBV RNA and an uninterrupted DR in the liver biopsies of 2 patients that had been collected within 1 year of clearance of serum HBsAg, suggesting the possibility of ongoing low-level replication.
In our previous study, none of these patients had appreciable HBV RNA demonstrable by in situ hybridization,8 and the low-level viral transcription was only detected in this study using the more sensitive method of RT-PCR. In a similar RT-PCR study, Fong et al. were unable to completely eliminate all the HBV DNA from the RNA preparations to adequately determine HBV transcription.11 However, we overcame this problem by increasing the quantity of DNAse 1 and temperature of digestion in excess of the recommendations of the manufacturers (Gibco BRL, Grand Island, NY), even though this markedly diminished the total amount of RNA (data not shown). Therefore, we were able to specifically demonstrate the presence of HBV RNA in this population at the expense of diminished sensitivity.
The seminested PCR methodology using primers spanning the human HBV direct repeat region was not able to detect Dane particle DNA as predicted. However, PCR products were observed using positive-control HBV-infected patients’ liver samples harboring the CCC HBV-DNA replicative intermediate, as well as the PLC/PRF/5 cells, which produces HBsAg but not intact virions. Of note, one of seven integrated HBV-DNA sequences in the PLC/PRF/5 hepatoma cell line has an uninterrupted direct repeat region of HBV DNA.22 Therefore, the seminested PCR methodology of the direct repeat region is not specific for CCC HBV DNA, but two points should be borne in mind with regard to the likelihood of this assay detecting CCC HBV DNA versus integrated HBV DNA. First, the majority of HBV integrations described to date breach the direct repeat region as depicted in Fig. 1.20,22 Second, we were assessing DNA derived from the study patients’ hepatocytes, which may contain differing integration events in each cell as opposed to the single integrated intact direct repeat site found in every PLC/PRF/5 cell.
Another problem with the PCR assay of the direct repeat region may have been false-negative results. For example, it is probable that patient 7 had low-level HBV replication because he had detectable HBV RNA in the liver and HBV DNA in the serum (table 1). However, the negative DR region PCR suggests that he either lacked replicative intermediates within the liver or that this PCR methodology was unable to detect the CCC HBV DNA in this sample. It is conceivable that patient 7 had extrahepatic sites of viral replication to account for the detection of serum HBV DNA,15-17 but this hypothesis is less likely in view of the observation that this individual had a substantial amount of HBV RNA detected in his liver compared with the rest of the study group (Fig. 2B). Furthermore, our seminested DR region PCR methodology has a limited capacity to detect all subtypes of HBV, and therefore may have provided false-negative results for patient 7. Alignment studies of known subtype variations in the DR region reveal that the DRF1 and DRR2 primers will amplify all the known HBV subtypes, but the DRR3 primer lacks homology at the 3′ nucleotide with the ayrand adr subtypes, and therefore may not form an adequate 3′ clamp to amplify these HBV species.
In total, five of seven patients in our study group had evidence of either viral transcription or an intact direct repeat region of HBV DNA within the liver. However, only one of these subjects was determined as having HBV DNA in the serum by PCR,8 and therefore it is not known whether infectious HBV particles were actually being synthesized and exported from hepatocytes. Cases of HBV replication have been documented in HBsAg-negative subjects, but these patients differed from our study group because they had evidence of continued hepatitis with elevated serum aminotransferases, and liver histology consistent with chronic hepatitis.28,29 In these studies, more exhaustive methods were employed to assess the ability of HBV to replicate by either inoculating chimpanzees with potentially infected tissue or by cloning, sequencing, and/or transfecting the persistent HBV DNA into hepatoma cell lines.28,29 However, it was beyond the scope of the present study to employ these procedures, which are not practical for assessing multiple patient samples.
There is other histological, immunologic, and clinical evidence to suggest that latent HBV retains the ability to produce proteins and replicate in the liver of patients who have cleared serum HBsAg. Despite the improvement in liver histology of patients who clear serum HBsAg, the majority of subjects who have been studied still retain a mild degree of hepatitis.1,10,11 This finding may be partially explained by the detection of vigorous T-cell responses and antibody reactivity at remote intervals after clinical recovery from both acute and chronic HBV infection.30,31 Although it is still a matter for debate, the maintenance of immunity after resolution of disease may be dependent on the ongoing antigenic stimulation by persistent viral proteins.31 The converse also appears to be true, in that the maintenance of active humoral and cellular immune responses may be important for preventing recurrent disease.31
There is considerable data documenting reactivation of HBV infection in immunosuppressed individuals to support the hypothesis that HBV is undergoing low-level replication in the liver following the clearance of serum HBsAg. For example, there are several reports of de novo Hepatitis B in liver-transplant recipients who received their liver grafts from HBsAg-negative but anti-HBc- and/or anti-HBs-seropositive donors.4,6,7,32 Reactivation of chronic HBV infection has been observed in HBsAg-negative patients undergoing renal transplantation who either had a remote history of HBV infection or received a kidney from an HBsAg-negative donor with serological evidence of previous HBV infection.3,5,7 All these reports of reactivated infection in HBsAg-negative patients have occurred in the setting of chemotherapy or immunosuppression, which have the known adverse effects of reducing immune surveillance and stimulating viral transcription, as well as replication, via the glucocorticoid-response element in the HBV genome.33
In summary, this study suggests that persistent HBV replication occurs in the liver many years following HBsAg clearance from the serum. The production of complete virions within the liver provides an explanation for the vigorous immune responses to HBV proteins many years after resolution of the disease, the potential for reactivation of hepatitis in immunocompromised individuals, and the passage of infection from HBsAg-negative liver-transplant donors with a remote history of HBV infection. Moreover, it is possible that this low-level HBV replication may be another cofactor for the development of hepatocellular carcinoma in HBsAg-negative patients, in addition to cirrhosis and the integration of HBV DNA into the host’s genome. Finally, the demonstration of low-level HBV replication within the liver following resolution of hepatitis raises the question whether extrahepatic sites may also support viral replication after loss of serum HbsAg.
Footnotes
Acknowledgement: The authors are grateful to Dr. Graeme Alexander for critical review of the manuscript, Dr. Jerome Zeldis for help with the study design and provision of the HBsAg PCR primers, and Dr. Richard Sallie for supplying the albumin PCR primers.
Abbreviations: HBsAg, Hepatitis B surface antigen; HBV, Hepatitis B virus; PCR, polymerase chain reaction; RT, reverse transcription; CCC, covalently closed circular.
Received July 22, 1997; accepted February 17, 1998.
Address reprint requests to: Robert P. Perrillo, M.D., Section of Gastroenterology and Hepatology, Ochsner Medical Institutions, 1520 Jefferson Highway, New Orleans, LA 70121. Fax: (504) 842-7466.
Copyright © 1998 by the American Association for the Study of Liver Diseases.


